您好,欢迎来到半岛电竞官方网址 ! [
登录
] [
免费注册
]
设为首页
手机站
m.cnreagent.com
扫一扫,直接在手机上打开
客户服务
会员服务
广告服务
关键词服务
成功案例
联系我们
网站导航
半岛bd体育手机客户端
企业
资讯
字典
首页
半岛游戏平台官网入口网址 设备
试剂耗材
机械设备
化工原料
半岛电子游戏官方网站
通用试剂
标准品
检测试剂
试剂盒
生物试剂
核酸
细胞
抗体
蛋白
基因与染色体
纳米材料
材料与耗材
半岛游戏平台官网入口网址
分析半岛游戏平台官网入口网址
实验室常用设备
光学半岛游戏平台官网入口网址 及设备
物性测试半岛游戏平台官网入口网址 及设备
测量/计量半岛游戏平台官网入口网址
环境监测半岛游戏平台官网入口网址
生命科学半岛游戏平台官网入口网址 /设备
行业专用半岛游戏平台官网入口网址
工业在线及过程控制半岛游戏平台官网入口网址
半岛游戏平台官网入口网址 设备制造
配件、耗材与服务
传感器
工业系统
连接技术
工业电源
IO-Link
安全技术
机械设备
泵阀
半岛游戏平台官网入口网址 仪表
成型设备
传质设备
制冷设备
压力容器
成套设备
真空设备
制药设备
包装设备
环保设备
储运设备
分离设备
传热设备
干燥机械
混合设备
输送设备
辅助设备
粉碎设备
反应设备
塑料专用设备
涂料专用设备
橡胶专用设备
其它设备
原料
原料、中间体
香精香料
农用化学品
医药与生物化工
气体
日化原料
功能化学品
参展厂商
资讯
专题
资料
会展
品牌
招聘
发布求购
化学试剂
中间体
标准品
试剂盒
化工原料
分析半岛游戏平台官网入口网址
实验室设备
物性测试
环境监测
生命科学
耗材配件
机械设备
热点资讯
大连化物所发现通过调控电解液中水含量可实现锌离子电池的高容量和长寿命
工信部等八部门印发《推进磷资源高效高值利用实施方案》
生态环境部正式批复陕煤千亿煤化工项目
5万吨!又一光伏制造项目落户新疆
经历供需两弱后全球化学品2024年将复苏,欧洲仍疲软
68548-08-3, 艾迪科 ADK STAB LA-82
BT系列分配/流量型智能蠕动泵
ViscoQC 100-H旋转粘度计
Genie G 10超纯水系统
库伦法阳极液
Snoop检漏液
SG23-FK-CN便携式多参数测试仪
HYDRANAL卡式炉水标
1,6-己二醇二丙烯酸酯
碳十二|90622-57-4
Celfinder 核酸染料
对苯二甲酸
MA100C 红外水份测定仪
羟甲基纤维素
防风精密电子天平
羟乙基纤维素
dPette+电动移液器
钙荧光探针
厂商动态&技术
那么如何检测狗狗是否感染了细小病毒呢
客户感谢信|深耕合作之壤,盛放共赢之花
欧美克半岛游戏平台官网入口网址 软件质量体系,实现全周期智能管理
南京景竹生物2024年春节放假安排通知
寓教于乐|提升员工环保意识,促进企业绿色发展
修订玩具国际标准ISO 8124-5研讨会在昆山顺利举办
热搜
专场
粉碎设备
色谱半岛游戏平台官网入口网址
纯化设备
包装设备
生物试剂
检测试剂
推荐半岛bd体育手机客户端
[半岛bd体育手机客户端 ]
PAL-3便携式数显折射仪(宽量程&高精度)
[半岛bd体育手机客户端 ]
亚硫酸盐(总SO2,酶法分析)检测试剂盒
[半岛bd体育手机客户端 ]
HYDRANAL-卡式炉水标(150-160℃)
[半岛bd体育手机客户端 ]
附着力检测仪(拉开法)
[半岛bd体育手机客户端 ]
硅酸镁锂Laponite XLG-XR
[半岛bd体育手机客户端 ]
氟表面活性剂Capstone? FS-30
[半岛bd体育手机客户端 ]
SH系列数显式推拉力计
[半岛bd体育手机客户端 ]
电子布氏硬度计HBE-3000A
[半岛bd体育手机客户端 ]
BT101S调速型蠕动泵(DG6×4)
[半岛bd体育手机客户端 ]
(S)-(-)-环己基丙氨酸甲酯盐酸盐
[半岛bd体育手机客户端 ]
XY-100MW-T智能卤素水分测定仪
[半岛bd体育手机客户端 ]
COD-571化学需氧量测定仪
[半岛bd体育手机客户端 ]
ACQUITY UPLC HSS C18色谱柱, 100?, 1.8μm, 2.1 mm×100 mm
[半岛bd体育手机客户端 ]
Abbemat 3000折光仪
[半岛bd体育手机客户端 ]
入门级烟气分析仪testo 310
[半岛bd体育手机客户端 ]
HALOX? 350有机缓蚀剂
[半岛bd体育手机客户端 ]
GBS-5000B玻璃珠灭菌器
[半岛bd体育手机客户端 ]
农药残留速测仪TMYQ-108P
[半岛bd体育手机客户端 ]
50L旋转蒸发仪N-53
[半岛bd体育手机客户端 ]
银铜,箔, Ag85Cu15, Hard Temper, 0.007mm厚
[半岛bd体育手机客户端 ]
TS-3D摇床
[半岛bd体育手机客户端 ]
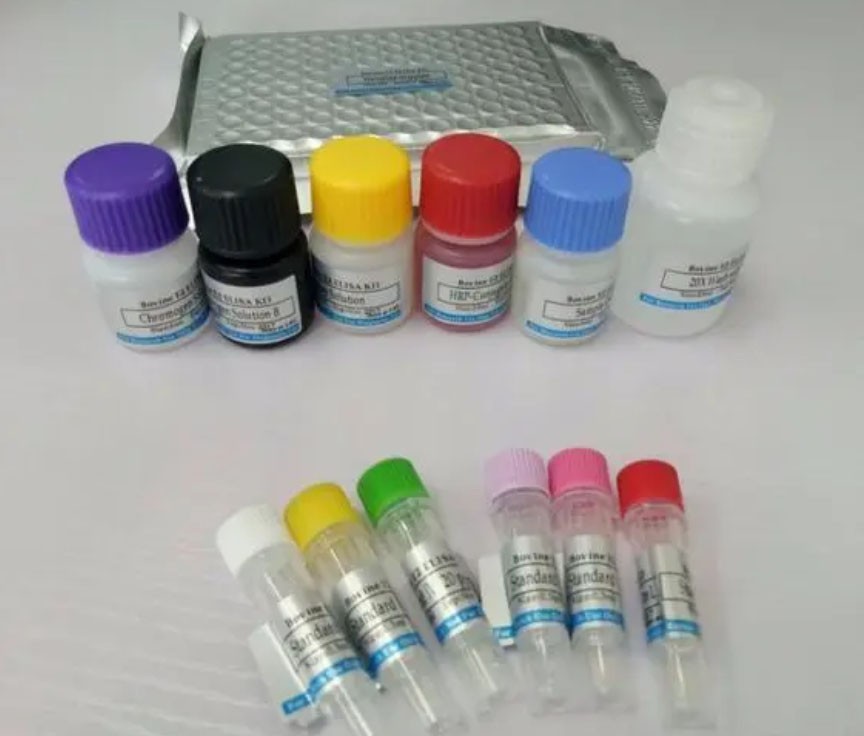
犬巴贝斯虫PCR检测试剂盒
推荐供应商
梯希爱(上海)化成工业发展有限公司
北京百灵威科技有限公司
Strem Chemicals INC.
阿法埃莎(中国)化学有限公司
西格玛奥德里奇中国有限公司
上海谷研实业有限公司
广东翁江化学试剂有限公司
天元航材(营口)科技股份有限公司
杭州天迈生物科技有限公司
埃朗科技国际贸易(上海)有限公司
赛多利斯(SARTORIUS)半岛游戏平台官网入口网址
多条一次性半岛bd体育手机客户端 新线投产,赋能生物制药供应链再升级
赛多利斯完成对诺华赛色谱板块的收购
无菌工艺盛宴——“无菌工艺保障技术交流会”,新年重磅袭来!
Quintix? 分析天平
Cubis? 高性能称重天平
昆山美美超声半岛游戏平台官网入口网址
超声波清洗器常见故障分析
超声波清洗机的日常维护及保养
超声波清洗机换能器常见问题
KM-700VDE-2中文液晶台式双频超声波清洗器
KQ-250旋钮型台式超声波清洗器
其林贝尔(Kylin-Bell)半岛游戏平台官网入口网址
脱色摇床TS-200使用说明
其林贝尔半岛游戏平台官网入口网址 大事记
旋涡混合器的工作原理和选购指南
脱色摇床TS-2型
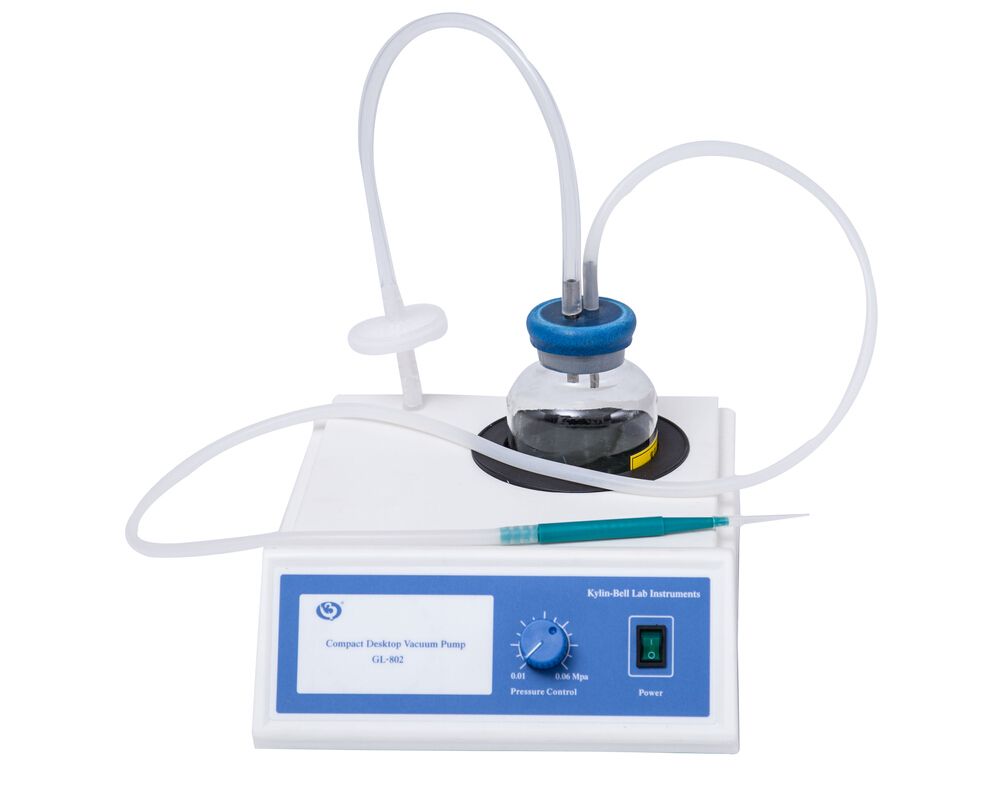
微型台式真空泵GL-802型
雷弗(LEADFLUID)蠕动泵
液体粘稠对蠕动泵传输影响
蠕动泵和计量泵的区别
蠕动泵停用后的维护措施
BT100S调速型智能蠕动泵(YZ15)
BT100F分配型智能蠕动泵(YZ25×2)
推荐半岛bd体育手机客户端
CD19分子(CD19)ELISA检测试剂盒
Bante321-CL便携式氯离子浓度计
RYJ-600ZG3全自动热压机,500℃,双平板
PH818笔式PH计
T1热电偶合金,线, Ni90Cr10, 0.025mm直径, Annealed Temper
MN 660硅藻土滤纸,150 mm
ClO2+型二氧化氯测量计
粮食水分仪(实用型)MC-7825G
全自动固体密度计DH-120N
UV-1200紫外可见分光光度计
THX-2005低温恒温循环器
SMN-R反射率仪
试剂
SNOOP检漏液
伟伯科技
咨询
缓冲液Technical pH 4.01
梅特勒-托利多
咨询
T1热电偶合金,线, Ni90Cr10, 0.025mm直径, Annealed Temper
Goodfellow
咨询
D/L-乳酸快速检测试剂盒
伟伯试剂盒
咨询
2-哌啶乙基氯盐酸盐
翁江试剂
咨询
呕吐毒素快速检测试剂盒(20分钟)
华安麦科
咨询
助剂原料
ADK STAB LA-82
伟伯科技
咨询
LORD 7650聚氨酯粘合剂
伟伯工业品
咨询
三氧化二硼
天元航材
咨询
铜材抗氧化剂
众事达表面处理
咨询
紫外线吸收剂UV-531(BP12)
南京米兰化工
咨询
均苯四甲酸
常熟联邦
咨询
半岛游戏平台官网入口网址
SK-O330-Pro圆周型摇床
大龙兴创半岛游戏平台官网入口网址
咨询
DYCP-40C型半干式碳板转印电泳仪
广州伟伯科技
咨询
快速水份测定仪HS153
梅特勒-托利多
咨询
颚式粉碎仪BB 50
弗尔德半岛游戏平台官网入口网址
咨询
WT300S大扭矩调速型蠕动泵(YT25×2)
雷弗流体科技
咨询
testo 610迷你型温湿度仪
德图半岛游戏平台官网入口网址
咨询
机械设备
FDS 高效超细砂磨机
上海法孚莱机电
咨询
JDS-1型数控电动击实仪
南京土壤半岛游戏平台官网入口网址
咨询
YMPZ-2自动金相试样磨抛机
金相机械
咨询
YKA型圆块孔式石墨换热器
科兴石墨设备
咨询
恒温恒湿培养箱(升级款)LHP-300E
三发科学半岛游戏平台官网入口网址
咨询
PHN 10 纳米销棒式砂磨机
派勒集团
咨询
企业招聘
濮阳豫沪不锈钢制造有限公司
不锈钢导压管
河南 濮阳
陕西谱光微视科技有限公司
科研助理和技术人员
陕西 西安
浙江康聚药业有限公司
研发人员合成(五险一金+双休+年终奖)
浙江 绍兴
青岛金美达贸易有限公司
销售代表
山东 青岛
上海纳洛捷生物科技有限公司
销售工程师 技术工程师
江苏 苏州
上海京诚检测技术有限公司
销售业务经理
上海 浦东
深圳贝尔信息科技有限公司
中级软件开发工程师 base 深圳
广东 深圳
东莞市亿富机械科技有限公司
电气工程师
广东 东莞
浙江智瑞自控阀门有限公司
销售经理
浙江 温州
湖北荆洪生物科技股份有限公司
操作工、成本会计、人事主管
湖北 襄阳
留言求购
【宝予德】我对您的Genex单道移液器半岛bd体育手机客户端 感兴趣,请发具体的信息及报价,谢谢!
【芯倍科仪】我对您的5CWY-D编码器半岛bd体育手机客户端 感兴趣,请发具体的信息及报价,谢谢!
【品享科技】我对您的PN-PLTF卫生纸饱满度测定仪半岛bd体育手机客户端 感兴趣,请发具体的信息及报价,谢谢!
【威福光电】我对您的手提式色差仪WR-18半岛bd体育手机客户端 感兴趣,请发具体的信息及报价,谢谢!
【德诚信环保】我对您的GRH-56蒸汽型加湿器半岛bd体育手机客户端 感兴趣,请发具体的信息及报价,谢谢!
【宝品科技】我对您的实验室双螺杆挤出机半岛bd体育手机客户端 感兴趣,请发具体的信息及报价,谢谢!
【深圳新产业生物】我对您的全自动化学发光仪MAGLUMI 800半岛bd体育手机客户端 感兴趣,请尽快发具体的信息及报价,谢谢!
【川思(transene)】我对您的金蚀刻剂TFA半岛bd体育手机客户端 感兴趣,请发具体的信息及报价,请问可以刻蚀PET材料上的金膜吗,目标在PET板上镀0.2um的金,0.03um的铬做粘附层,该蚀刻试剂对PET有损害吗
[求购]
甲基丙烯酸2-(叔丁基氨基)乙酯 10吨
启源(广东)医药化工有限公司
[求购]
Orion 1816DO纯水溶氧表
广州伟伯科技有限公司
[求购]
求购氧化镁 20吨
南京瑞之祥贸易有限公司
[求购]
(多罐)振动球磨仪 1台
广州远想医学生物技术有限公司
[求购]
罗硝唑 1公斤
上海固康生物科技有限公司
会展中心
2024第二十届中国南京科学半岛游戏平台官网入口网址 及实验室装备展览会
南京国际展览中心
2024.03.21
2024第二十届中国南京教育装备暨科教技术展览会
南京国际展览中心
2024.03.21
2024第12届华中科教半岛游戏平台官网入口网址 与技术装备展览会
武汉·中国光谷科技会展中心
2024.03.26
2024成都国际分析与测试、生化诊断技术、实验室设备展览会
成都世纪城新国际会展中心
2024.03.28
2024国际(广州)涂料工业展览会
广州保利世贸博览馆
2024.05.15
2024中国(广东)国际医疗制造与设计创新展览会
保利世贸博览中心
2024.05.31
2024宁波化工新材料、新科技、新装备展览会
宁波国际会展中心
2024.06.28
2024年世界机器人大会(北京)展览会
北京亦创国际会展中心
2024.08.15
2024(第二十一届)中国国际化工展览会
上海新国际博览中心
2024.09.19
第五届高比能固态电池关键材料技术大会
昆山·美仑国际酒店
2024.01.18
半岛电竞官方网站首页
细胞房培养基过滤真空泵选择
AP-5复合物β亚基ELISA试剂盒操作注意事项
定量包装秤出现电磁吸合力下降的原因
微波消解仪安全使用指南
大鼠小肠粘膜组织提取物操作流程
兔淋巴细胞功能相关抗原3ELISA试剂盒操作步骤
小鼠D-乳酸elisa检测试剂盒试验时,注意事项
小鼠脂肪酸合成酶(FASN)检测试剂盒试剂配制
实验室安装元素分析仪需注意哪些问题?
行业百科
锂电材料
荧光微球体检测
叶绿素仪/叶绿素测定仪
PCR试剂
热空气消毒箱
石墨烯材料
化学发光分析仪
塑料助剂
表面活性剂(原料)
资料中心
化工字典
常用临床生化名词辞汇中文、英文名
化学品MSDS数据查询
化学结构式图库
元素周期表
实验室常用设备维护与保养——pH计
流动相中缓冲盐使用方法与注意事项
溶解氧的检测方法与影响因素
二氧化碳培养箱在细胞培养过程中如何避免污染
企业在生产过程中产生的一般固体废物该如何处置?
香料香精
粽子香精
刺楸
氨草油
杜荆叶油
嘉陵花
淀粉
地笋
白千层烯
窃衣
安古树酮
名企推荐
伟伯科技
上海谷研
翁江试剂
天元航材
希玛仪表
岛津
Goodfellow
梅特勒-托利多
是德科技
安捷伦科技
北京锐志汉兴
安东帕
赛多利斯
提赛环科半岛游戏平台官网入口网址
广州一思通
英国百灵达
林上半岛游戏平台官网入口网址
舜宇恒平半岛游戏平台官网入口网址
大龙兴创半岛游戏平台官网入口网址
科导超声半岛游戏平台官网入口网址
雷弗流体科技
苏净安泰
上海仪电雷磁
加野科学半岛游戏平台官网入口网址
哈希水质分析
上海沪析
奥豪斯
科尔诺电子
兰泰半岛游戏平台官网入口网址
泰斯特半岛游戏平台官网入口网址
索光光电
三信仪表
山度半岛游戏平台官网入口网址
三恩时科技
上海亚荣
前视红外光电
格林凯瑞半岛游戏平台官网入口网址
锐智科电
安莱立思半岛游戏平台官网入口网址
安妙半岛游戏平台官网入口网址
杭州爱华半岛游戏平台官网入口网址
安柏精密半岛游戏平台官网入口网址
个性关键词
最新半岛bd体育手机客户端
耐驰·闪射法导热仪
雷弗流体·蠕动泵
沃特世色谱柱ACQUITY UPLC HSS PFP
淀粉酶检测试剂盒
恒平精密电子天平
雷磁·水质多参数分析仪
苏净安泰·洁净工作台
台式超声波清洗器
恒温恒湿培养箱
大龙仪·器磁力搅拌器
乐枫Rephile·纯水系统
安东帕·旋转粘度计
梅特勒便携式电导率仪
MS 3000粒度分析仪
赛多利斯·手动移液器
石油半岛bd体育手机客户端 多功能测定仪
TSI(提赛环)塑料分析仪
百灵达·溶解氧测量计
哈希水质浊度仪
生物安全柜生物检测仪
欧美克粒度分析仪
施魏科·研磨机
SNOOP 检漏液
自动化吸头(适配Cybio设备)
色谱科·离子载体
离子交换树脂
Capstone氟表面活性剂
荧光试剂·脂性Cy5
卡尔费休试剂HYDRANAL
加野·尘埃粒子计数器
电子密度天平
便携式气体检测仪
S10无线蓝牙pH计
手持电池测试仪
泰斯特·蒸馏水器
BCA蛋白定量试剂盒
山度·推拉力计
锚杆拉拔仪
雄发半岛游戏平台官网入口网址 ·密度计
自动脱盖离心机
BINDER·恒温恒湿箱
杭州爱华·声级计
Ace pressure tube
棕榈酸甲酯
半岛游戏平台官网入口网址 铭牌标签
过孔滑环
接近开关
TRACE 1600气相色谱仪
20加仑泄漏应急处理桶套装(通用)
通用型吸油栅
DS1000钻石吸嘴易拉装,960个(10*96)
DS200ST钻石吸嘴易拉装,灭菌型,960个(10*96)
DS1000ST钻石吸嘴易拉装,灭菌型,960个(10*96)
网状气瓶柜
骨形成蛋白1(BMP1)ELISA检测试剂盒
细胞色素P450家族成员1A2(CYP1A2)ELISA检测试剂盒
弗林蛋白酶(FUR)ELISA检测试剂盒
围脂滴蛋白5(PLIN5)ELISA检测试剂盒
四旋蛋白1(TSPAN1)ELISA检测试剂盒
蛋白酶激活受体4(PAR4)ELISA检测试剂盒
基质金属蛋白酶19(MMP19)ELISA检测试剂盒
G蛋白偶联受体109B(GPR109B)ELISA检测试剂盒
晶状体蛋白βB2(CRYbB2)ELISA检测试剂盒
细胞色素P450家族成员2B6(CYP2B6)ELISA检测试剂盒
溶质载体家族30成员4(SLC30A4)ELISA检测试剂盒
水通道蛋白8(AQP8)ELISA检测试剂盒
环指蛋白39(RNF39)ELISA检测试剂盒
嘌呤能受体P2Y14(P2RY14)ELISA检测试剂盒
层粘连蛋白α3(LAMa3)ELISA检测试剂盒
多萜醇二磷酸寡糖蛋白环糊精糖基转移酶(DDOST)ELISA检测试剂盒
SPARC相关组件钙结合蛋白1(SMOC1)ELISA检测试剂盒
序列相似家族20成员A(FAM20A)ELISA检测试剂盒
MYC关联因子X(MAX)ELISA检测试剂盒
骨形成蛋白6(BMP6)ELISA检测试剂盒
组织型纤溶酶原激活因子(tPA)ELISA检测试剂盒
失调蛋白3(ATXN3)ELISA检测试剂盒
动力蛋白胞浆2重链1(DYNC2H1)ELISA检测试剂盒
沉默调节蛋白7(SIRT7)ELISA检测试剂盒
丫叉同源物2(SSH2)ELISA检测试剂盒
兴奋性氨基酸转运蛋白5(EAAT5)ELISA检测试剂盒
桥连整合因子2(BIN2)ELISA检测试剂盒
ATP结合盒转运蛋白C9(ABCC9)ELISA检测试剂盒
Ⅲ型前胶原羧基端原肽(PIIICP)ELISA检测试剂盒
S100钙结合蛋白B(S100B)ELISA检测试剂盒
X-射线修复交叉互补蛋白1(XRCC1)ELISA检测试剂盒
连接附着分子3(JAM3)ELISA检测试剂盒
纤维胶凝蛋白2(FCN2)ELISA检测试剂盒
含EGF样模块粘蛋白样激素受体1(EMR1)ELISA检测试剂盒
CD27结合蛋白(CD27BP)ELISA检测试剂盒
半乳糖凝集素2(GAL2)ELISA检测试剂盒
发动蛋白1样蛋白(DNM1L)ELISA检测试剂盒
可卡因安非他明调节转录肽(CART)ELISA检测试剂盒
CREB调节转录辅激活因子3(CRTC3)ELISA检测试剂盒
烟碱型胆碱受体α5(CHRNa5)ELISA检测试剂盒
B-细胞淋巴瘤因子9(Bcl9)ELISA检测试剂盒
细胞色素P450家族成员2E1(CYP2E1)ELISA检测试剂盒
透明质酸合酶1(HAS1)ELISA检测试剂盒
鸟氨酸脱羧酶抗酶1(OAZ1)ELISA检测试剂盒
磷酸肌醇-3-激酶催化亚基δ肽(PIK3Cd)ELISA检测试剂盒
防御素β103A(DEFb103A)ELISA检测试剂盒
前胶原赖氨酸-2-酮戊二酸-5-双加氧酶1(PLOD3)ELISA检测试剂盒
UDP-葡萄糖糖蛋白糖基转移酶1(UGGT1)ELISA检测试剂盒
白介素12受体β2(IL12Rb2)ELISA检测试剂盒
Sestrin 2蛋白(SESN2)ELISA检测试剂盒
衔接因子相关蛋白复合体2μ1(AP2m1)ELISA检测试剂盒
亨廷顿关联蛋白1(HAP1)ELISA检测试剂盒
丝裂原激活蛋白激酶12(MAPK12)ELISA检测试剂盒
非受体型蛋白酪氨酸磷酸酶6(PTPN6)ELISA检测试剂盒
ATP结合盒转运蛋白C8(ABCC8)ELISA检测试剂盒
Notch同源物2(NOTCH2)ELISA检测试剂盒
2',5'-寡腺苷酸合成酶样蛋白(OASL)ELISA检测试剂盒
蛋白激酶Bα(PKBa)ELISA检测试剂盒
胞裂蛋白12(SEPT12)ELISA检测试剂盒
防御素α6(DEFa6)ELISA检测试剂盒
1-[5-(4-fluorophenyl)-1H-pyrazol-4-yl]-N-methylmethanamine
2-Cycloheptylethanamine
{[4-(TRIFLUOROMETHYL)PHENYL]PHENYLMETHYL}PIPERAZINE 2HCL
N,N-Dimethyl-7H-purin-6-amine
5-(2-cyclohexylethyl)-4-ethyl-4H-1,2,4-triazole-3-thiol
4-AMINO-3,5-DIMETHYLPYRIDINE1-OXIDE
(R)-2-(3-Phenylpropyl)pyrrolidine-2-carboxylic acid hydrochloride
Ethyl 2-amino-4-(2-chlorophenyl)thiophene-3-carboxylate
1-(3-Bromophenyl)piperidin-2-one
N-Ethyl-4-fluoroaniline
ETHYL 4-(PHENYLSULFONYLMETHYL)-3,4-DIHYDRO-2H-BENZO[B][1,4]OXAZINE-2-CARBOXYLATE
2-AMINO-4-(BENZYLOXY)-5-METHOXYBENZONITRILE
3-Oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-5-carboxylic acid
Phenyl(pyridin-4-yl)methanone
4,4'-Sulfonylbis(bromobenzene)
1-(Cyclohex-1-en-1-yl)piperidine
METHYL 2-(3-(2-METHOXYBENZYL)UREIDO)BENZOATE
5-Chloro-3-(chloromethyl)-1,2,4-thiadiazole
3'-Fluoro-[1,1'-biphenyl]-2,4'-dicarboxylic acid
(S)-2-(2-Amino-4-methylpentanamido)acetic acid
2-(2-Fluoropyridin-3-yl)propan-2-ol
6-Hydrazinylquinoline hydrochloride
Benzoyl chloride, 5-bromo-2-iodo-
1-piperidin-4-ylpent-4-en-1-one hydrochloride
N-(4-fluoro-3-methylphenyl)-2,2-dimethylpropanamide
Boc-4-benzoyl-D-phenylalanine
1,3-Di(pyridin-2-yl)urea
11-Aminoundecanoic acid
5-(4-Nitrophenyl)-1H-tetrazole
9-Methoxy-7H-furo[3,2-g]chromen-7-one
N-(2-CHLORO-5-NITROPHENYL)BENZAMIDE
2-(4-Chlorophenyl)-2-[4-(4-methylphenyl)piperazin-1-yl]acetic acid
N-(2-aminoethyl)cyclobutanecarboxamide
(S)-Benzyl 3-methylpiperazine-1-carboxylate
1,5-Dichloropentan-3-one
N-Isopropylcyclohexanamine
3-Bromophenyl cyclopropyl ketone
3-(PYRAZIN-2-YLOXY)-BENZYLAMINE HCL
7-amino-1,3-dimethyl-5-(2-methylphenyl)-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6-carbonitrile
2-(2,4-Dichlorobenzyl)-1H-benzo[d]imidazole
1-Aminocyclopropanecarboxamide
Platinum(IV) chloride
1-Phenyl-3-(trifluoromethyl)-1H-pyrazole-4-carboxylic acid
Ethyl 2-cyano-2-methylpropanoate
1-ETHYL-1,3-BENZODIAZOLE-5-CARBONITRILE
3-(2,4-Dihydroxyphenyl)propanoic acid
cis-4-Methyl-cyclohexylamine hydrochloride
1-BOC-4-(3-ETHOXYCARBONYL-PYRIDIN-2-YL)-PIPERAZINE
1256346-01-6, 3-Isopropoxy-5-(trifluoromethoxy)phenylboronic Acid
N-{[(2-sec-butylphenyl)amino]carbonothioyl}-2-iodobenzamide(7421943)
1707594-42-0, 1-(pyrrolidin-3-yl)-1H-1,2,3-triazole-4-carboxamide
38231-86-6, 6-Amino-4-Hydroxymethyl-Cyclohex-4-Ene-1,2,3-Triol
50290-51-2, 2-Methylpyrimido[1,2-a]benzimidazol-4(1H)-one
1160247-14-2, tert-butyl2-carbamoyl-6-azaspiro[2,5]octane-6-carboxylate
1445950-94-6, tert-butyl6-(hydroxymethyl)-2-azaspiro[4,4]nonane-2-carboxylate
1219981-37-9, 2-[Methyl(tetrahydro-2H-pyran-4-ylmethyl)amino]-nicotinic acid
774158-28-0, 4-(chloromethyl)-2-cyclopentyl-1,3-thiazole
89267-43-6, 5-(Ethoxycarbonyl)-2-(hydroxymethyl-6-methyl-4-(3-nitrophenyl)nicotinic Acid(Nitrendipine Impurity)
169124-63-4, (1r,5s,6s)-rel-8-azabicyclo[3,2,1]octan-6-ol
1189878-73-6, 1-(1,3-Thiazol-4-yl)ethanamine
846032-02-8, 7-(4-(4-(naphthalen-1-yl)piperazin-1-yl)butoxy)-3,4-dihydro-1,8-naphthyridin-2(1H)-one
2090315-88-9, 1-(3-methyloxetan-3-yl)propan-1-one
6515-18-0, 1-(2-Methoxymethoxy-phenyl)ethanone
1105192-67-3, (6-Oxo-3-pyridin-3-ylpyridazin-1(6{H})-yl)acetic acid
14964-98-8, Bancroftinone
426842-71-9, 4-Bromo-3-chloro-6-methoxyquinoline
显示更多
显示较少
合作伙伴:
粉体网
广州伟伯
旗下网站:
试剂信息网
化工原料网
半岛游戏平台官网入口网址 设备网
机械设备网
map





 m.cnreagent.com
m.cnreagent.com